Huntington’s Disease (HD) Research: Key Targets and Antibody Tools
Huntington’s disease (HD) was first systematically described in 1872 by the American physician George Huntington, after whom the condition is named. It is a fully penetrant autosomal dominant neurodegenerative disorder — inheriting just one mutant allele is sufficient to cause disease. A hallmark feature is genetic anticipation, where successive generations typically develop symptoms earlier than their parents. Global prevalence is approximately 5–10 per 100,000, with slightly lower rates in East Asian populations, though diagnostic awareness has risen sharply in recent years. Classic symptoms include uncontrollable choreiform movements, psychiatric disturbances, and progressive cognitive decline. Onset is usually between 30 and 50 years of age, with a disease course of 10–25 years ending in complications such as dysphagia, pneumonia, or cardiac failure.
The root cause of HD is an abnormal expansion of CAG trinucleotide repeats in exon 1 of the HTT gene ; 36–39 repeats show reduced penetrance; and ≥40 repeats are almost fully penetrant. The expanded CAG tract encodes an abnormally long polyglutamine (polyQ) stretch at the N-terminus of Huntingtin, conferring toxic gain-of-function: misfolding and aggregation, transcriptional dysregulation, impaired axonal transport, blocked autophagy clearance, and suppressed BDNF production/transport. These events culminate in selective loss of medium spiny neurons (MSNs) in the striatum and cortical neurons.
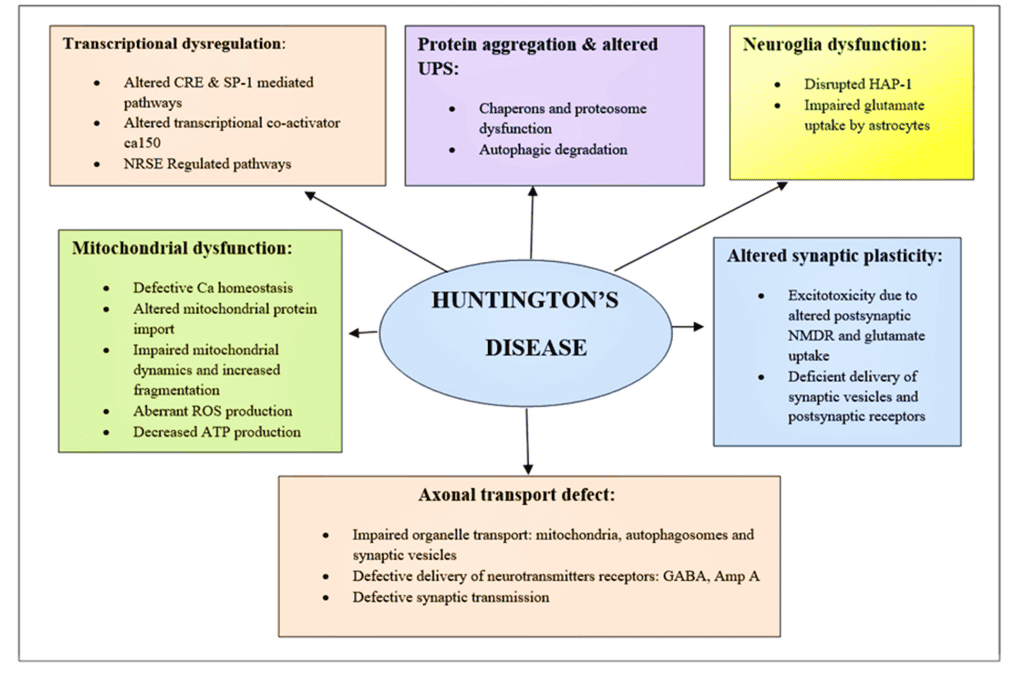
Fig 1.Schematic of diverging pathways leading to the pathogenesis of HD(doi.org/10.3390/brainsci12101389)
Key Targets in Huntington’s Disease Research
Although HD is caused by a single-gene mutation, its downstream pathology is extraordinarily complex, involving protein homeostasis, neurotrophic support, autophagy, excitotoxicity, and mitochondrial dysfunction. The table below summarizes the most widely accepted core targets and their roles in HD, providing a foundation for therapeutic intervention.
| Target |
Normal Biological Function |
Pathological Role in HD |
Research Applications |
| HTT (Huntingtin) |
Scaffold protein involved in vesicular transport, BDNF support, autophagy, and ciliogenesis |
polyQ expansion → toxic gain-of-function, aggregation, transcriptional dysregulation, impaired axonal transport |
mHTT aggregation ELISA, clearance assays, HTT-lowering validation |
| BDNF / TrkB |
Neurotrophic factor and receptor essential for striatal neuron survival and synaptic plasticity |
Mutant HTT represses BDNF transcription and axonal transport, causing neurotrophic deprivation |
BDNF ELISA, TrkB phosphorylation assays, neurotrophic rescue experiments |
| mTOR |
Master regulator of autophagy, protein synthesis, and cell growth |
Hyperactivation inhibits autophagy, exacerbating mHTT accumulation and neuronal injury |
Autophagic flux assays, mTOR inhibitor screening |
| GRIN2B (NMDAR) |
NMDA receptor subunit critical for synaptic transmission and plasticity |
Extrasynaptic overactivation triggers excitotoxicity and calcium overload |
Calcium imaging, excitotoxicity models, NMDAR antagonist screening |
Therapeutic Implications: Direct HTT lowering (ASO, RNAi, gene editing), BDNF-TrkB pathway activation, mTOR modulation to restore autophagy, and selective blockade of extrasynaptic NMDAR represent the most promising disease-modifying strategies today.

Figure 2. Integrated schematic of Huntington’s disease pathogenesis: mutant Huntingtin toxic gain-of-function, aggregation, BDNF-TrkB loss, mTOR-autophagy impairment, excitotoxicity, and other key nodes (Integrated HD pathogenesis pathways). Antibodies enable node-specific analysis.
Latest Research Progress
The table below summarizes major clinical and basic research advances in HD from leading journals, trial registries, and authoritative sources.
| Theme |
Key Findings/Trial Results |
Publication/Update |
Potential Impact |
Citation |
| AMT-130 (uniQure AAV-miHTT) |
Phase I/II 36-month interim analysis: high-dose group showed 75% slowing of disease progression on cUHDRS (P=0.003), CSF NfL below baseline, acceptable safety |
September 2025 |
First HD gene therapy demonstrating clear disease-modifying potential; possible one-time treatment milestone |
[1] |
| Tominersen (Roche) |
GENERATION HD2 adjusted after interim review to continue only 100 mg arm; higher-dose/frequency arms previously terminated due to risk/benefit concerns |
2025 update |
Exploring non-allele-selective HTT lowering in younger, lower-burden patients |
[2] |
| WVE-003 (Wave Life Sciences) |
First allele-specific ASO; SELECT-HD early data show ~40–50% mHTT reduction with preserved wild-type HTT; correlated with slower caudate atrophy |
2025 data |
Clinical proof-of-concept for allele-specific approaches, expanding precision therapy to broader population |
[3] |
Current Research Challenges in Huntington’s Disease (as of November 2025)
Despite encouraging signals from AMT-130 and others, no disease-modifying therapy is approved yet. Major hurdles include lack of sensitive early biomarkers, brain delivery and durability issues, allele-selective vs non-selective safety/efficacy trade-offs, ongoing somatic CAG expansion, insensitive clinical endpoints, high cost/access barriers, and pathway complexity limiting single-target interventions. These challenges underscore the need for multi-pathway, systems-level research — precisely the rationale behind abinScience’s BDNF-TrkB, mTOR, and NMDAR tool portfolio.
Below are abinScience’s latest recombinant proteins and antibodies targeting core HD pathways. Catalog numbers link directly to product pages.
Why Choose abinScience HD Research Tools? (Click to expand)
- Covers key HD downstream pathways: BDNF-TrkB neurotrophic support, mTOR-autophagy regulation, NMDAR-mediated excitotoxicity
- InVivoMAb in vivo-grade antibodies (low endotoxin) for long-term dosing in mouse, rat, and NHP HD models
- Multicolor flow antibodies (APC/FITC/PE/PerCP) enable striatal neuron subpopulation sorting and functional analysis
- All products undergo rigorous QC for batch-to-batch consistency and high activity — build reliable HD in vitro/in vivo models faster
| Type |
Catalog # |
Product Name |
| Protein |
MB935012 |
Recombinant BDNF Protein, N-His |
| HB935012 |
Recombinant Human BDNF Protein, N-His |
| HB935022 |
Recombinant Human BDNF Protein, N-His |
| HC330012 |
Recombinant Human GRIN2B Protein, N-His |
| HW733012 |
Recombinant Human MTOR Protein, N-His |
| HW733022 |
Recombinant Human MTOR Protein, N-His |
| HS892012 |
Recombinant Human TrkB /NTRK2 Protein, N-His |
| Antibody |
HS892016 |
Research Grade Anti-Human TrkB /NTRK2 (ZEB85) |
| HB935014 |
Anti-BDNF Polyclonal Antibody |
| HC330014 |
Anti-GRIN2B Polyclonal Antibody |
| HB935107 |
Anti-Human BDNF Antibody (SAA0471) |
| HB935137 |
Anti-Human BDNF Antibody (SAA0471), APC |
| HB935117 |
Anti-Human BDNF Antibody (SAA0471), FITC |
| HB935127 |
Anti-Human BDNF Antibody (SAA0471), PE |
| HB935147 |
Anti-Human BDNF Antibody (SAA0471), PerCP |
| HW733013 |
Anti-Human MTOR Antibody (SAA2296) |
| HW733014 |
Anti-Human MTOR Polyclonal Antibody |
| HS892107 |
Anti-Human TrkB /NTRK2 Antibody (SAA0477) |
| HS892137 |
Anti-Human TrkB /NTRK2 Antibody (SAA0477), APC |
| HS892117 |
Anti-Human TrkB /NTRK2 Antibody (SAA0477), FITC |
| HS892127 |
Anti-Human TrkB /NTRK2 Antibody (SAA0477), PE |
| HS892147 |
Anti-Human TrkB /NTRK2 Antibody (SAA0477), PerCP |
| HS892014 |
Anti-TrkB /NTRK2 Polyclonal Antibody |
| HB935010 |
InVivoMAb Anti-Human BDNF (Iv0093) |
abinScience: Empowering Huntington’s Disease Research with High-Quality Recombinant Proteins and Precision Antibodies
References
- Tong H, Yang T, Xu S, et al. Huntington’s Disease: Complex Pathogenesis and Therapeutic Strategies. International Journal of Molecular Sciences. 2024;25(7):3845. doi:10.3390/ijms25073845
- Piao X, Li D, Liu H, Guo Q, Yu Y. Advances in Gene and Cellular Therapeutic Approaches for Huntington’s Disease. Protein & Cell. 2025;16(5):307–337. doi:10.1093/procel/pwae042
- Estevez-Fraga C, Tabrizi SJ, Wild EJ. Huntington’s Disease Clinical Trials Corner: March 2024. Journal of Huntington’s Disease. 2024;13(1):1–14. doi:10.3233/JHD-240017
- Farag M, Tabrizi SJ, Wild EJ. Huntington’s Disease Clinical Trials Update: September 2024. Journal of Huntington’s Disease. 2024;13(4):409–418. doi:10.1177/18796397241293955
- Sampaio C. Huntington Disease – Update on Ongoing Therapeutic Developments and a Look Toward the Future. Parkinsonism & Related Disorders. 2024;122:106049. doi:10.1016/j.parkreldis.2024.106049
- Azman KF, Zakaria R. Brain-Derived Neurotrophic Factor (BDNF) in Huntington’s Disease: Neurobiology and Therapeutic Potential. Current Neuropharmacology. 2025;23(4):384–403. doi:10.2174/1570159X22666240530105516
For research use only. Not for use in diagnostic or therapeutic procedures.

 中文
中文 English
English 한국어
한국어 日本語
日本語 Español
Español Français
Français Русский
Русский


